双氧水(H₂O₂)是一种绿色氧化剂,其氧化产物仅为水(H₂O)和氧气(O₂),无二次污染,被广泛应用于化工合成(如环氧丙烷制备)、环境保护(污水深度氧化处理)、医疗消毒(伤口清洁、器械灭菌)、农业生产(病虫害防治、根系供氧)等领域。据统计,全球双氧水年需求量超千万吨,且随着 “绿色化工” 理念普及,需求仍在持续增长。然而,目前工业上 95% 以上的双氧水通过蒽醌法生产,其核心流程为 “烷基蒽醌氢化→氢蒽醌氧化→双氧水萃取→蒽醌循环”,该技术存在三大关键问题:1.高能耗与高污染。反应需在特定温度(40-60℃)、压力(0.3-0.5MPa)下进行,依赖化石能源驱动;2.催化剂与原料局限。氢化步骤依赖贵金属钯(Pd)催化剂,成本高昂且资源稀缺,而且反应过程需消耗氢气(H₂),而 H₂多来自化石燃料制氢(如天然气重整),并非 “全生命周期绿色”;3.安全与运输风险:双氧水化学性质不稳定(浓度>30% 时易分解爆炸),传统蒽醌法需 “集中生产-长距离运输”,需使用防爆容器和低温运输设备,不仅增加物流成本,还存在泄漏、爆炸等安全隐患。因此,传统双氧水合成技术存在显著瓶颈,无法满足当前 “低能耗、低污染、高安全” 的发展需求,这为光催化绿色合成技术的研发提供了核心驱动力。
光催化绿色双氧水合成技术的核心原理是:光催化剂吸收太阳光后产生电子-空穴对,电子(e⁻)将氧气(O₂)还原为超氧自由基(・O₂⁻),进而质子化生成双氧水(H₂O₂);空穴(h⁺)则氧化水(H₂O)生成质子(H⁺)和双氧水。整个过程仅需水、氧气和太阳光,无需高温高压、有机溶剂或贵金属(可采用非金属催化剂如石墨相氮化碳 g-C₃N₄或其他聚合物材料),天然具备 “绿色、低成本、安全” 的优势,完美匹配双氧水合成的 “全生命周期绿色化” 需求,然而目前光催化绿色双氧水合成的实际应用受到太阳能到双氧水的能量转换效率低、光催化剂结构稳定性差、双氧水浓度低等问题的制约。针对以上问题,北京大学深圳研究生院环境与能源学院周鹏助理教授课题组与北京大学材料科学与工程学院郭少军教授课题组、环境科学与工程学院童美萍教授课题组展开合作,在2023-2025年期间开发了一系列高性能的聚合物光催化剂材料,用于直接将太阳能、水、空气高效且稳定地转变为高浓度的双氧水,相关成果分别发表在《Nature Synthesis》、《Nature Communications》(三篇)、《Journal of the American Chemical Society》、《Angewandte Chemie International Edition》期刊上,为光催化双氧水合成技术提供了从催化剂结构设计到反应机制的系统性解决方案,推动了低碳与绿色化学在新能源领域的发展。
1. 五配位的镓单原子光合成双氧水
针对光催化双氧水合成过程中两电子水氧化势垒高的问题,在氮化碳光催化剂表面设计了一种新型五配位的镓单原子助催化剂(Ga-N5)(图1a-c),这种新型的高配位Ga-N5位点不仅提升了氮化碳光催化剂的光生载流子分离效率,还可以促进表面决速的两电子水氧化生产双氧水的过程。在可见光驱动下,CNIO-GaSA的过氧化氢产率达到368.5μmol h-1(图1d),是未经Ga原子位锚定氮化碳催化剂的4倍。在相同反应条件下(光强、温度、反应体积等),CNIO-GaSA催化水氧气合成过氧化氢的质量活性达331.7μmol g-1 h-1,高于当时已报道的光催化剂(图1e)。CNIO-GaSA产过氧化氢的转换效率为0.4%,远高于自然界植物光合作用的能量转化效率(~0.1%)。理论分析表明,高配位的Ga-N5位点可以通过同时形成Ga-O与N-O键来大幅度降低两电子水氧化过程中羟基中间体的形成势垒(图1f),不同于未改性的氮化碳材料通过独立的N-O键来稳定水氧化产生的羟基中间体(图1g-h)。该研究以“Photocatalysis of water into hydrogen peroxide over an atomic Ga-N5 site”为题发表在Nature Synthesis期刊上,北京大学博士后谭浩与周鹏助理教授为论文共同第一作者,北京大学郭少军教授为论文通讯作者1。

图1.(a)CNIO-GaSA的合成路径;(b)CNIO-GaSA的高分辨透射电镜照片;(c)镓元素的X射线吸收边曲线;(d)不同材料光合成双氧水的时间-浓度曲线;(e)CNIO-GaSA与已报道材料的活性比较;(f)CNIO-GaSA与氮化碳材料产生双氧水的反应势垒;(g)氮化碳材料表面与中间体的差分电荷密度图;(h)CNIO-GaSA材料表面与中间体的差分电荷密度图
2. 三配位的间隙铝单原子促进光合成双氧水反应中氮化碳的层间电荷转移效率
当前用于光催化双氧水合成的材料主要为二维层状的聚合物材料,包括氮化碳与共价有机骨架材料(Covalent Organic Frameworks,COF)。由于二维层状的聚合物材料中层与层之间为分子间作用力(图2a),导致层与层之间的电荷转移效率低,极大地限制了聚合物光催化剂材料生产双氧水的效率。鉴于此,团队在氮化碳材料的层间引入了三配位的间隙铝原子(Al-N3)(图2b),该间隙铝原子可以通过3s、3p轨道杂化同时与相邻的氮化碳层形成化学键作用(图2c),加强层间的电荷转移。此外,Al-N3桥式位点能够活化临近的表面氮原子,降低两电子水氧化生产H2O2的反应势垒(图2d)。由于这些优势,团队实验合成了富含Al-N3结构的CNNT-Al材料(图2e-f),在光催化反应合成H2O2的过程中获得了1410.2 μmol g-1 h-1的质量活性,性能超过了所有CN光催化剂,比自然界植物的太阳能转化为化学能的效率(~0.1 %)高7倍,达到了0.73 %。最为重要的一点是,CNNT-Al构筑的流动相反应器能够稳定工作200 h,制备的H2O2可以直接用于降解水体中的污染物(图2g)。该研究以“Al-N3 Bridge Site Enabling Interlayer Charge Transfer Boosts the Direct Photosynthesis of Hydrogen Peroxide from Water and Air”为题发表在Journal of the American Chemical Society期刊上,北京大学博士后谭浩与周鹏助理教授为论文共同第一作者,北京大学郭少军教授为论文通讯作者2。

图2.(a)氮化碳材料的层间电荷转移;(b)CNNT-Al的层间电荷转移;(c)氮化碳与CNNT-Al的PDOS图;(d)不同间隙原子的两电子水氧化势垒图;(e)CNNT-Al的透射电镜图;(f)CNNT-Al的X射线吸收谱图与晶体结构;(g)CNNT-Al的长期反应活性与装置图
3. 分子内极化调控COF光催化剂材料合成双氧水的性能
相比于氮化碳光催化剂,二维层状的COF光催化剂因为其结构性质的可调性在光催化双氧水合成中受到了最多的关注。然而,由于传统COF材料内部的光生电荷分离效率较低,因此限制了其太阳能到化学能的转换效率。对此,团队通过在COF材料中引入适量的苯基作为电子给体,成功制备出一种含有电子供体-受体对的COF光催化剂材料(图3a),在不使用牺牲剂的情况下,实现了水、空气和太阳光直接光催化产生H2O2。其中COF-N32材料具有最佳的分子内极性,平衡了电荷分离效率与材料的稳定性,在水溶液中获得了高效且稳定的H2O2产率(605 μmol g-1 h-1)和0.31%的光化学转换效率(图3b),为同期COF材料的最高性能(图3c),而且具有较高的量子效率与稳定性(图3d-e)。该研究以“Covalent organic frameworks for direct photosynthesis of hydrogen peroxide from water, air and sunlight”为题发表在Nature Communications期刊上,北京大学博士后刘福洋与周鹏助理教授为论文共同第一作者,北京大学童美萍教授为论文通讯作者3。

图3.(a)COF-N31、COF-N32、COF-N33的结构图;(b)不同材料的活性对照;(c)COF-N32与已报道材料的活性比较;(d)COF-N32在不同波长下的量子效率;(e)COF-N32材料的活性稳定性
4.多重电荷转移通道的构建及其对COF光催化剂合成双氧水的促进效应
虽然通过构筑电子供体-受体对可以在一定程度上提升COFs材料的光生电荷分离效率,但是难以满足更高效率的反应需求。鉴于此,团队采用双氰基官能化调控COFs的电荷转移通道数以促进激子分离与转移,并降低COFs中两电子水氧化的能量势垒,实现了太阳光下利用水与空气即可高效光合成H2O2,产生的H2O2溶液能够用于水体净化。其中具有缺电子特性的氰基基团可作为COFs中额外的电子受体,增加了COFs中供体到受体的电荷转移通道数。相比于COF-0CN(具有2条电子转移通道)和COF-1CN(具有3条电子转移通道),COF-2CN(具有4条电子转移通道)展现出更好的激子解离与转移行为(图4a-c),从而提高了电荷分离效率。相比于COF-0CN、COF-1CN及传统光催化剂(g-C3N4和P25),COF-2CN在纯水中具有更高的H2O2产量(1601 μmol g-1 h-1)(图4d),为同期COF材料的最高性能(图4e),而且在量子效率与稳定性方面也表现突出(图4f-g)。此外,COF-2CN也能在连续流反应器和自然太阳光照射下的放大反应器中持续高效地光合成H2O2,其产生的H2O2溶液能够高效灭活抗生素抗性细菌(图4h-i)。该研究以“Efficient Photosynthesis of Hydrogen Peroxide by Cyano-Containing Covalent Organic Frameworks from Water, Air and Sunlight”为题发表在Angewandte Chemie International Edition期刊上,北京大学博士后侯阳辉与周鹏助理教授为论文共同第一作者,北京大学童美萍教授为论文通讯作者4。
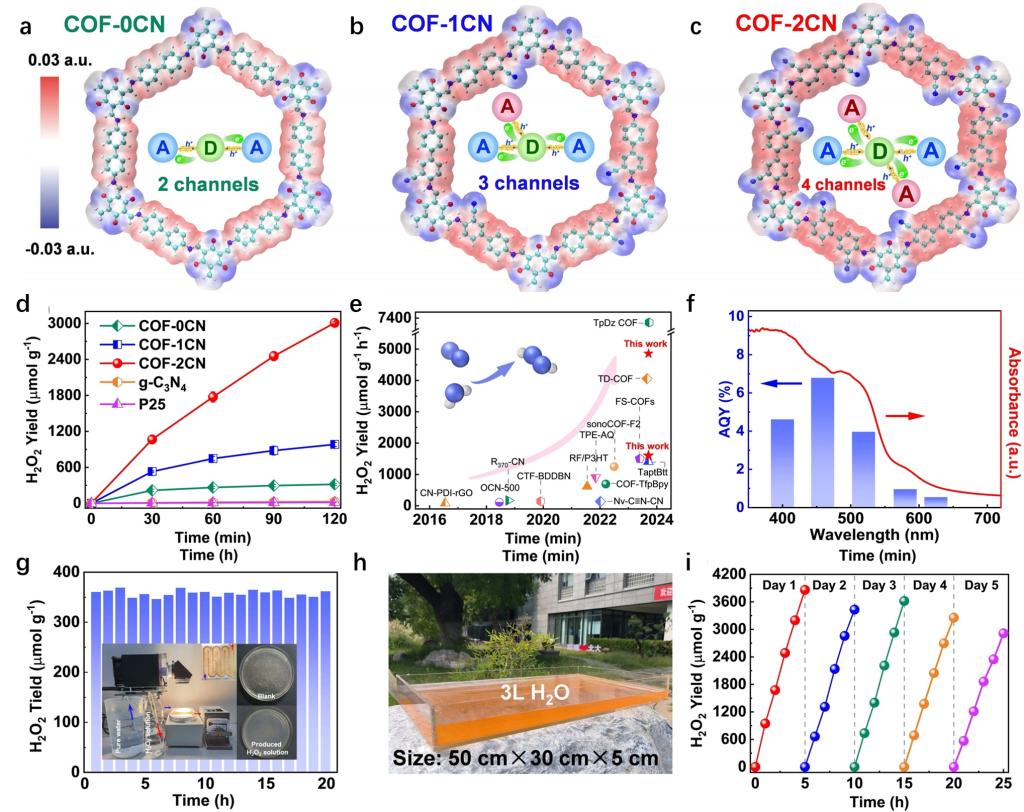
图4. (a-c)COF-0CN、COF-1CN、COF-2CN的结构图与电荷转移通道;(d)不同材料随时间的活性对比;(e)COF-2CN与已报道材料的活性比较;(f)COF-2CN在不同波长下的量子效率;(g)COF-2CN的长期反应活性与装置图;(h)COF-2CN的户外装置图;(i)COF-2CN在户外反应中的长期反应活性
5. 刚性COFs材料的构建及其稳定的光合成双氧水性能
在光催化水与氧气反应合成双氧水的过程中,两电子的水氧化过程除了会限制COFs材料光合成双氧水的效率外,还会引起COFs材料的氧化与腐蚀,造成COFs材料的稳定性降低。针对该问题,团队通过一种简便的一锅法合成策略,将COFs材料中原有的可逆亚胺键直接转化为刚性的噻唑键图(5a-c),调节了骨架的π共轭与局部的电荷极化,这不仅促进了COFs材料的光生电荷分离,而且增强了COFs骨架的坚固性以及COFs材料在连续使用过程中的稳定性。更重要的是,具有最佳碳2p态的COFs(COF-S)中的噻唑键能有效地提高了相邻苯单元的活性,从而直接调节氧气吸附能垒并提高活性氧(ROS)的生成效率,使得其在不同的水体环境下仍旧对七种有毒新兴污染物具有出色的光催化降解效率(例如,仅需7分钟就能降解约99%的5毫克/升的对乙酰氨基酚)(5d-f),同时相比于传统的COF-A材料具备更高的稳定性(5g-h)。此外,COF-S可固定在连续流反应器和放大反应器中,在自然阳光照射下高效去除污染物(5i),这证明了其实际应用的可行性。该研究以“Rigid covalent organic frameworks with thiazole linkage to boost oxygen activation for photocatalytic water purification”为题发表在Nature Communications期刊上,北京大学博士后侯阳辉与周鹏助理教授为论文共同第一作者,北京大学童美萍教授为论文通讯作者5。
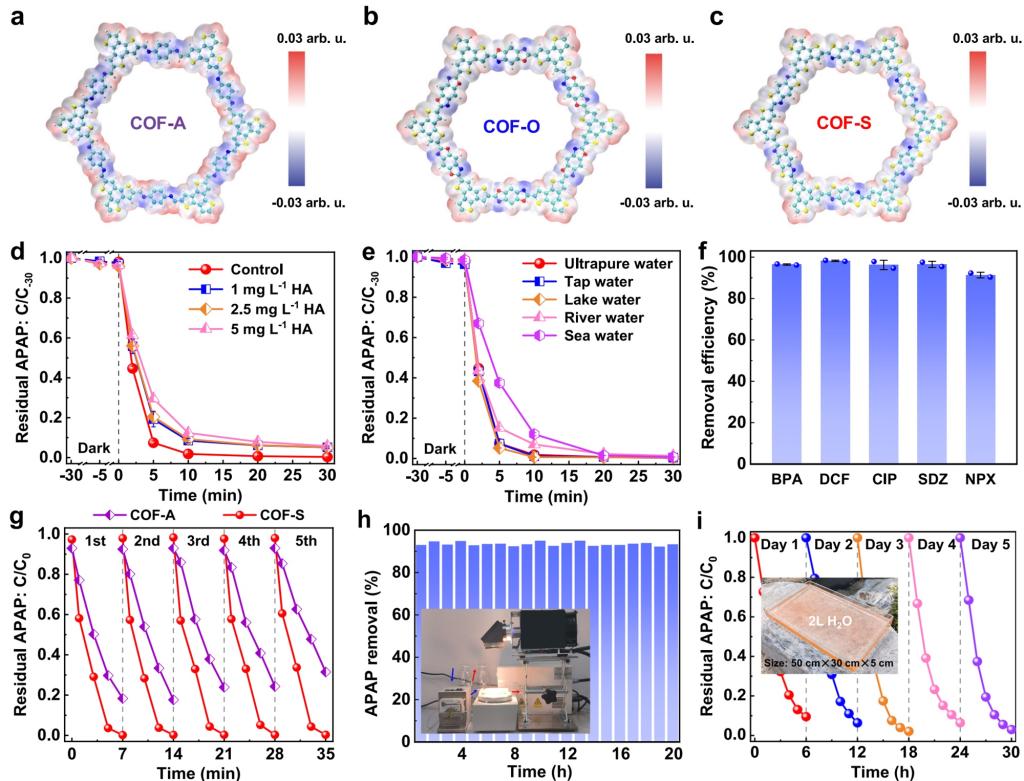
图5. (a-c)COF-A、COF-O、COF-S的结构图与电荷分布;(d)COF-S在不同HA浓度条件下降解对乙酰氨基酚的反应效率;(e)COF-S在不同水质条件下降解对乙酰氨基酚的反应效率;(f)COF-S对不同有机污染物的降解效率;(g)COF-S与COF-A的稳定性比较;(h)COF-S材料的长时间稳定性测试与反应装置;(i)COF-S材料的户外长时间稳定性测试与反应装置
6. 钽单原子催化剂促进光催化协同生成高浓度过氧化氢与高附加值生物质燃料
在光合成双氧水的过程中,两电子的水氧化步骤具有较高的反应势垒,成为了双氧水合成效率提升的主要障碍。虽然,大量的方法已经被开发用于降低两电子的水氧化势垒,但是目前报告的两电子水氧化势垒仍旧高于2 eV。鉴于此,团队利用相对低反应势垒的生物质脱氢反应替换了两电子的水氧化反应,同时结合低势垒的两电子氧气还原反应制备双氧水,该设计不仅可以大幅度降低了整体反应的能量势垒,而且可以同时产生高浓度的双氧水与高价值的生物质燃料。对此,团队研发了一种间苯二酚-甲醛树脂/碳负载的Ta-N2O2单原子催化剂(RF/C-TaSA)(图6a-c),能够利用易获取的茅草、松针和废纸高效地进行过氧化氢及高附加值化学品的人工光合作用合成(图6d-f)。通过结合催化剂创新和系统工程,构建了一个基于RF/C-TaSA的太阳能光催化装置,该装置可直接生产具有商业可行性的高浓度(3 wt%)过氧化氢以及高附加值化学品,且在70天的运行过程中无需耗能的分离过程(图6g-h)。此外,所生产的粗制浓缩过氧化氢还能轻松转化为固体过氧化氢粉末(Na2CO3·1.5H2O2),便于储存和运输,即使在6个月后仍具有高杀菌活性。该研究以“Separation-free artificial photosynthesis of concentrated hydrogen peroxide and value-added fuels over Ta atomic sites”为题发表在Nature Communications期刊上,北京大学博士后谭浩与周鹏助理教授为论文共同第一作者,北京大学郭少军教授为论文通讯作者6。
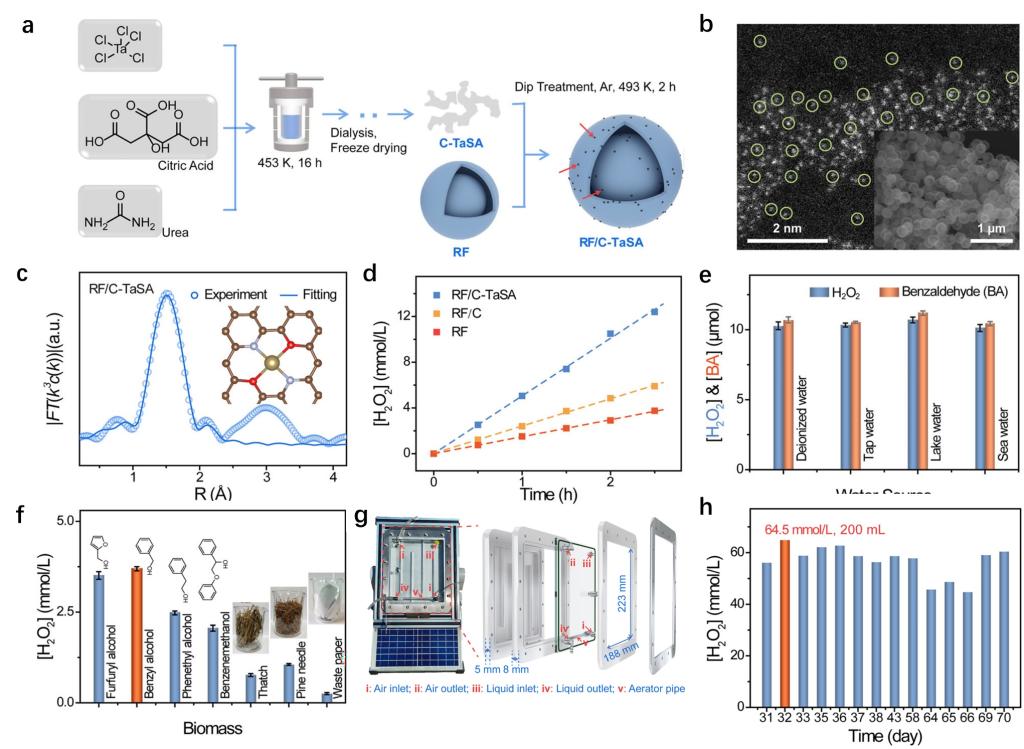
图6. (a)RF/C-TaSA的合成路径图;(b)RF/C-TaSA的透射电镜图;(c)RF/C-TaSA的扩展的X射线吸收谱与配位结构图;(d)RF/C-TaSA与其他材料的活性对照;(e)RF/C-TaSA在不同水体中合成双氧水的活性;(f)RF/C-TaSA在不同生物质存在条件下合成双氧水的活性;(g)RF/C-TaSA的户外反应装置;(h)RF/C-TaSA材料的户外长时间稳定性测试
以上研究工作得到了得到国家自然科学委海外高层次人才项目、深圳自然科学基金、北京大学深圳研究生院启动基金的资助,同时得到了曙光超级计算中心和上海同步辐射光源的支持。
论文链接:
1. Tan, H. #; Zhou, P.#; Liu, M.; Zhang, Q.; Liu, F.; Guo, H.; Zhou, Y.; Chen, Y.; Zeng, L.; Gu, L.; Zheng, Z.; Tong, M.; Guo, S., Photocatalysis of water into hydrogen peroxide over an atomic Ga-N5 site. Nat. Synth. 2, 557-563 (2023).
2. Tan, H. #; Zhou, P. #; Liu, M.; Gu, Y.; Chen, W.; Guo, H.; Zhang, J.; Yin, K.; Zhou, Y.; Shang, C.; Zhang, Q.; Gu, L.; Zhang, N.; Ma, J.; Zheng, Z.; Luo, M.; Guo, S., Al-N(3) Bridge Site Enabling Interlayer Charge Transfer Boosts the Direct Photosynthesis of Hydrogen Peroxide from Water and Air. J. Am. Chem. Soc 146, 31950-31960 (2024).
3. Liu, F. #; Zhou, P. #; Hou, Y.; Tan, H.; Liang, Y.; Liang, J.; Zhang, Q.; Guo, S.; Tong, M.; Ni, J., Covalent organic frameworks for direct photosynthesis of hydrogen peroxide from water, air and sunlight. Nat. Commun. 14, 4344 (2023).
4. Hou, Y. #; Zhou, P. #; Liu, F.; Lu, Y.; Tan, H.; Li, Z.; Tong, M.; Ni, J., Efficient Photosynthesis of Hydrogen Peroxide by Cyano-Containing Covalent Organic Frameworks from Water, Air and Sunlight. Angew. Chem. Int. Ed. 63, e202318562 (2024).
5. Hou, Y. #; Zhou, P. #; Liu, F.; Tong, K.; Lu, Y.; Li, Z.; Liang, J.; Tong, M., Rigid covalent organic frameworks with thiazole linkage to boost oxygen activation for photocatalytic water purification. Nat. Commun. 15, 7350 (2024).
6. Tan, H. #; Zhou, P. #; Gu, Y.; Liu, Y.; Chen, W.; Guo, H.; Lin, F.; Luo, H.; Cao, X.; Zeng, L.; Luo, M.; Guo, S., Separation-free artificial photosynthesis of concentrated hydrogen peroxide and value-added fuels over Ta atomic sites. Nat. Commun. 16, 8784 (2025).



